

ClustalX多重序列比对软件
v2.0 免费版- 软件大小:4.19 MB
- 更新日期:2020-08-25 15:27
- 软件语言:英文
- 软件类别:信息管理
- 软件授权:免费版
- 软件官网:待审核
- 适用平台:WinXP, Win7, Win8, Win10, WinAll
- 软件厂商:

软件介绍 人气软件 下载地址
ClustalX多重序列比对软件可以帮助用户分析生物数据,将生物序列数据添加到软件就可以执行分析,支持进行完全对准、只做引导树、从引导树进行对齐、重新排列所选序列、重新调整选定的残留范围、将轮廓2与轮廓1对齐、对齐引导树中的轮廓、将序列与配置文件1对齐等数据处理和分析方案,也可以结合软件提供的树形菜单处理序列,在计算树之前,必须在内存中有一个ALIGNMENT,可以使用FILE菜单的LOAD
SEQUENCES选项输入,或者您应该已经进行了完整的多重比对,并且比对仍在内存中,需要注意的是分析之前必须首先对序列进行对齐,新版添加了UPGMA算法以允许更快地构建树,用户现在可以选择使用邻居加入还是UPGMA。默认值仍然是NJ,但是用户可以通过设置clustering参数来更改此设置。

软件功能
CLUSTAL X(2.0
Clustal X是ClustalW多序列比对程序的Windows界面。它提供了一个集成的环境,用于执行多个序列和配置文件比对并分析结果。序列比对显示在屏幕上的窗口中。包含了通用的着色方案,可让您突出显示路线中的保守特征。窗口顶部的下拉菜单使您可以选择传统的多序列和谱图比对所需的所有选项。
您可以剪切和粘贴序列以更改比对的顺序。您可以选择要比对的序列子集;您可以选择要重新对齐的路线的子范围,然后重新插入到原始路线中。
可以进行比对质量分析,并且可以突出显示低分片段或异常残基。
ClustalX在Linux,Mac和Windows上可用。
序列输入
使用FILE菜单输入序列和概况(预先存在的比对术语)。无效的选项将被禁用。所有序列必须包含在1个文件中。自动识别7种格式:NBRF / PIR,EMBL / SWISSPROT,Pearson(Fasta),Clustal(* .aln),GCG / MSF(Pileup),GCG9 RSF和GDE平面文件。除用于指示GAP的“-”(MSF / RSF中的“。”)外,所有非字母字符(空格,数字,标点符号)都将被忽略。
序列/配置文件对齐
Clustal X具有两种模式,可以使用序列显示上方的开关进行选择:多重对齐模式和轮廓对齐模式。
要对一组序列进行多重对齐,请确保选择了多重对齐模式。然后显示单个序列数据区域。然后,ALIGNMENT菜单可让您生成用于对齐的引导树,或在引导树之后进行多重对齐,或进行完全多重对齐。
在PROFILE ALIGNMENT MODE中,显示两个序列数据区域,使您可以对齐2个比对(称为Profile)。配置文件还用于将新序列添加到旧比对中,或使用二级结构指导比对过程。旧版比对中的GAPS使用“-”字符表示。可以使用任何允许的格式输入个人资料;只需在每个间隙位置使用“-”(对于MSF / RSF,则使用“。”)。在配置文件对齐模式下,将显示“锁定滚动”按钮,您可以使用单个滚动条将两个配置文件一起滚动。当“锁定滚动”关闭时,两个配置文件可以独立滚动。
植物树
系统发育树可以从旧的比对(使用“-”字符读入以指示间隙)或在多次比对后仍显示比对的情况下计算得出。
对齐显示
比对显示在屏幕上,序列名称显示在左侧。序列比对仅用于显示,不能在此处进行编辑(通过剪切和粘贴序列名称来更改序列顺序除外)。
标尺显示在序列下方,从第一个残基位置的1开始(序列输入文件中的残基编号将被忽略)。
路线上方的线用于标记高度保守的位置。使用三个字符(“ *”,“:”和“。”):
“ *”表示具有单个完全保守残基的位置。
“:”表示以下“强”组之一是完全保守的:
STA
NEQK
NHQK
NDEQ
QHRK
MILV
MILF
HY
FYW
“。”表示以下“较弱”组之一是完全保守的:
CSA
ATV
SAG
STNK
STPA
SGND
SNDEQK
NDEQHK
NEQHRK
FVLIM
HFY
这些都是出现在Gonnet Pam250矩阵中的正得分组。强和弱组分别定义为强得分> 0.5和弱得分= <0.5。
对于轮廓对齐,如果在轮廓输入文件中找到任何数据,则在序列上方显示二级结构和空位罚分蒙版。
软件特色
对准质量分析
质量得分
Clustal X通过绘制比对的每一列的“保守分数”来指示比对的质量。高分表示列保存良好;低分表示低保守性。质量曲线绘制在路线下方。
还提供了两种方法来指示在比对中得分很差的单个残基或序列片段。
低分数残基由于其自然进化过程而稳定发散,因此预期在所有序列中均以中等频率出现。差异最大的序列可能具有最多的异常值。但是,突出显示的残基在指出序列错位时特别有用。请注意,突出显示的残基会聚在一起,强烈表明未对齐。这可能是由于多种原因引起的,例如:
由于对齐算法失败而导致的部分或全部未对齐。通常仅在困难的对齐情况下使用。
部分或全部错位,因为给定集合中的至少一个序列与其他序列部分或完全不相关。由用户检查序列集是否可比对。
蛋白质序列中的移码翻译错误会引起局部错配区域的突出显示。这些在数据库条目中非常常见。如果怀疑,则需要检查源DNA的3帧翻译。
有时,突出显示的残基可能指向具有某些生物学意义的区域。例如,如果蛋白质比对包含相对于主要序列集具有新功能的序列,则可能发生这种情况。重要的是在调用生物学解释之前,应排除其他解释,例如错误或序列的自然差异。
低分部
可以使用“低得分线段”选项突出显示路线中不可靠的区域。序列加权配置文件用于指示序列中评分不佳的任何片段。由于轮廓计算可能需要一些时间,因此提供了一个选项来计算低速分段。然后可以打开或关闭段显示,而不必重复耗时的计算。
有关低得分细分计算的详细信息,请参见下面的“计算”部分。
安装方法
1、打开clustalx.msi软件直接安装,点击next
2、提示软件的安装地址C:\Program Files (x86)\clustal\clustalx\
3、提示软件的安装准备界面,点击install
4、clustalx成功安装到电脑,点击finish结束安装
使用说明
1、点击 Load Sequences功能添加需要分析的序列,如果你有序列文件就加载
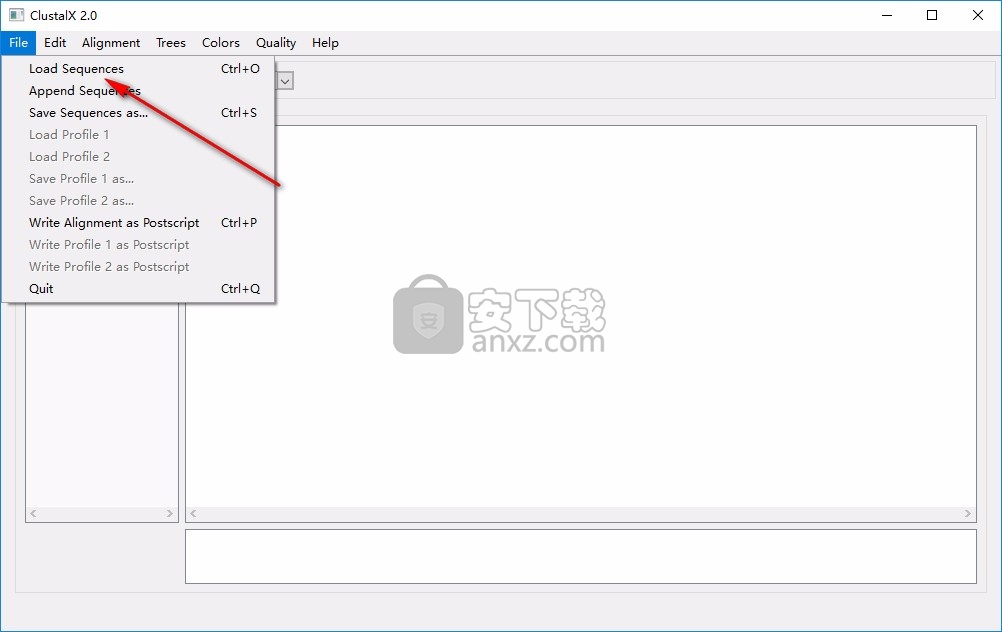
2、小编电脑没有序列文件所以就无法执行分析了,用户在分析的过程也可以选择追加序列
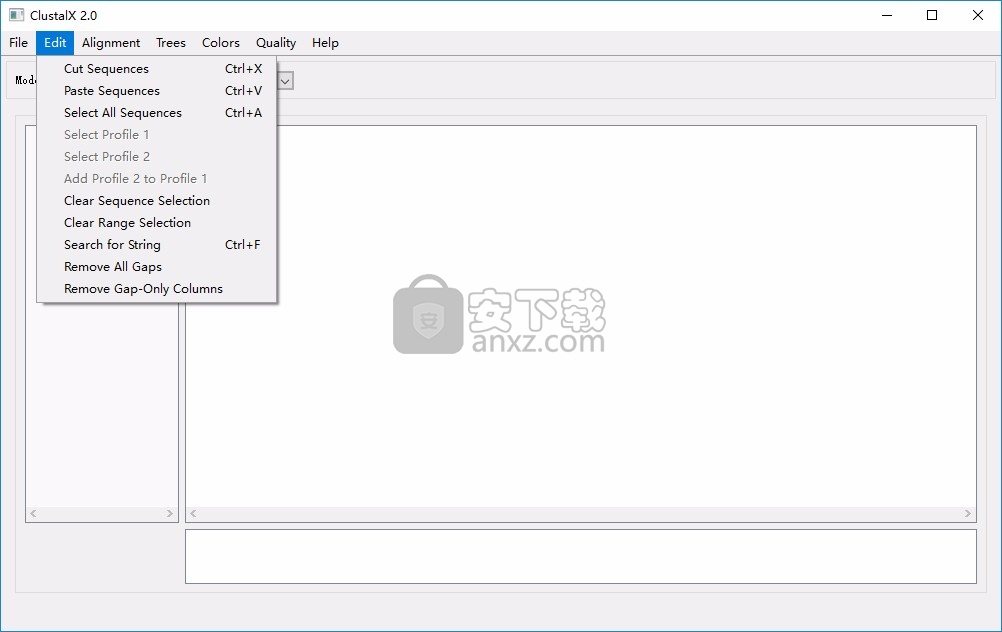
3、这里是软件的编辑功能,可以选择剪切序列、粘贴序列、选择所有序列、选择配置文件1、选择配置文件2
4、点击Alignment Parameters可以显示多种对齐方式,可以设置蛋白质数据对齐
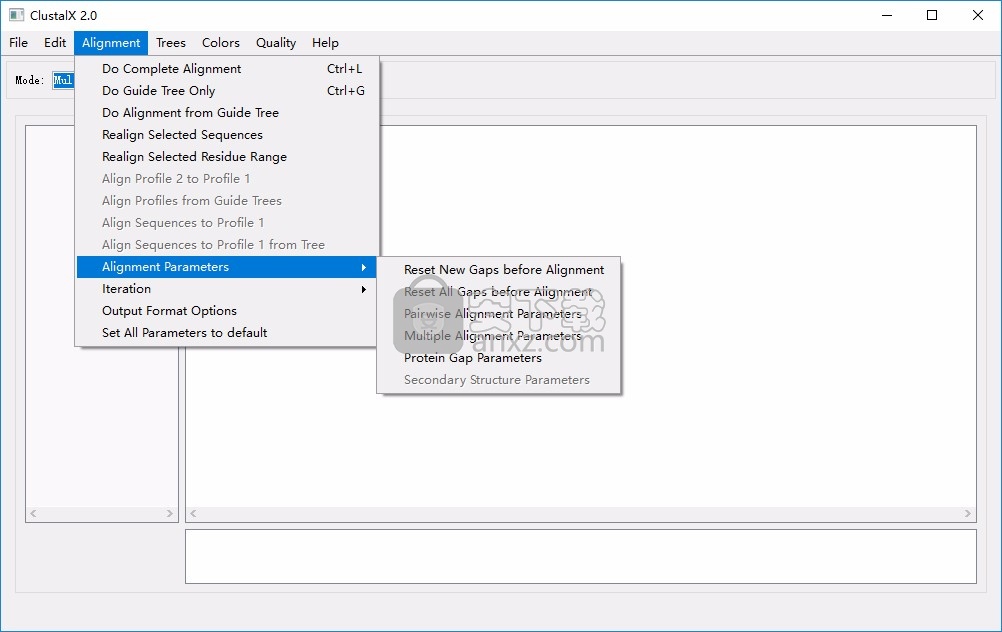
5、点击Iteration显示迭代内容,支持没有、重复每个对齐步骤、迭代最终对齐
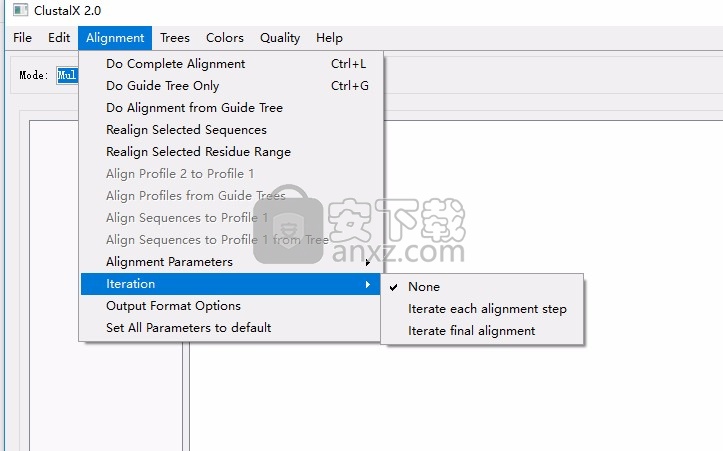
6、树形目录,支持 画树、Bootstrap N-J树、排除职位空缺、更正多个替代、输出格式选项、聚类算法
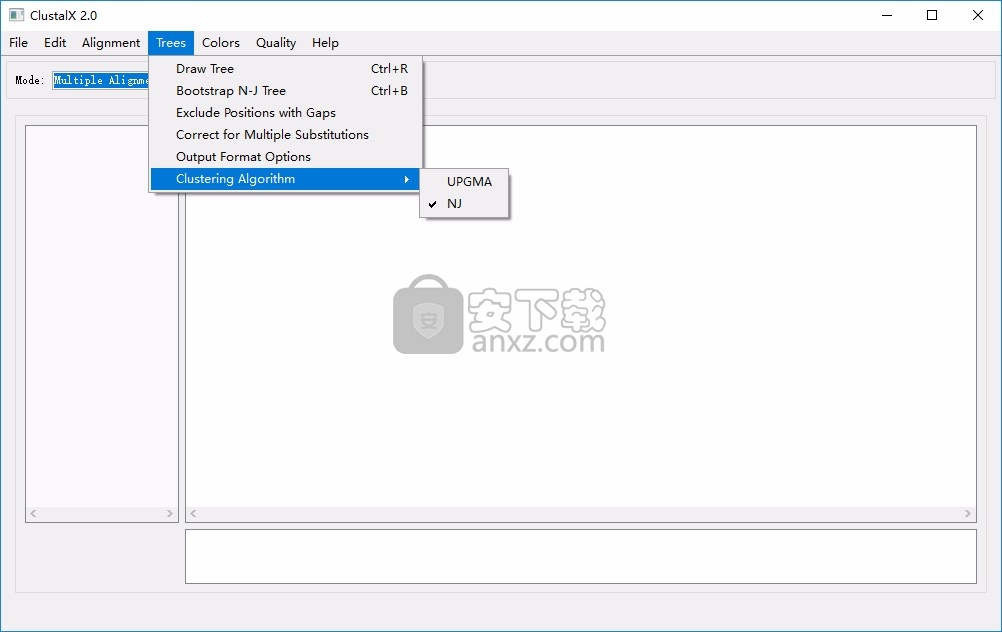
7、颜色设置功能,支持背景着色、黑和白、默认颜色、加载颜色参数文件
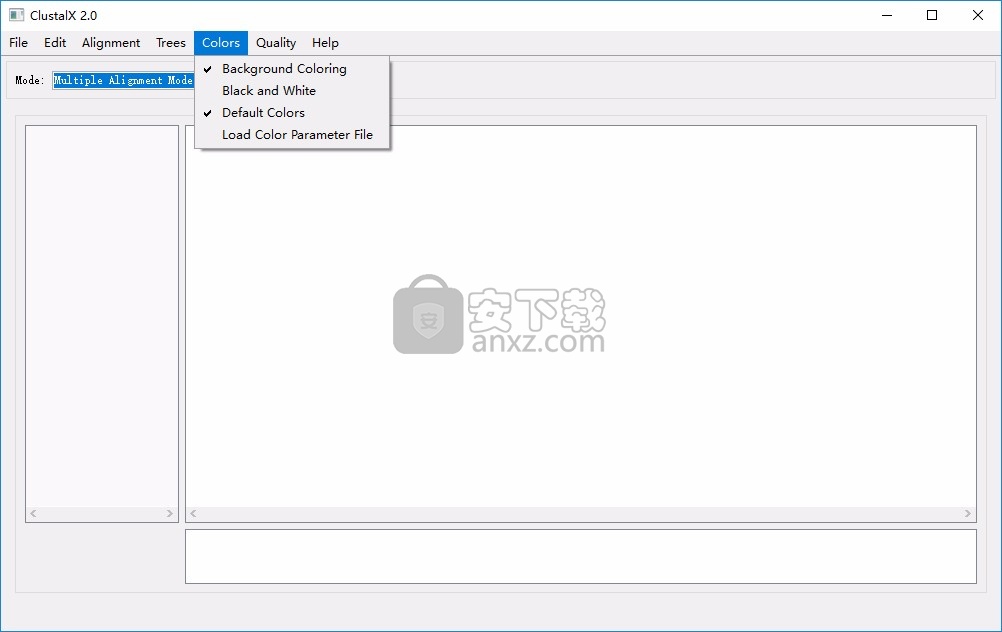
8、质量分析; 显示得分较低的细分、显示异常残基、低分段参数、列分数参数、将列分数保存到文件
9、更多的功能可以点击帮助查看软件相关的操作方式
官方教程
低分段参数
片段的最小长度:可以通过增加将要显示的片段的最小长度来隐藏短片段(甚至单个残基)。
DNA MARKING SCALE用于从突出显示的显示中删除不太重要的片段。增加比例以显示更多细分;减小比例以去除最低有效位。
蛋白质重量矩阵:评分表,描述每种氨基酸之间的相似性。该矩阵用于计算序列加权谱得分。提供了四种“内置”对数赔率矩阵:Gonnet PAM 80、120、250、350矩阵。当序列紧密相关时,仅对身份和最喜欢的保守替换给出高分的更严格的矩阵可能更适合。对于更发散的序列,使用“较软”的矩阵是合适的,该矩阵对许多其他频繁的替换给出较高的分数。此选项会自动重新计算得分较低的细分。
DNA权重矩阵:提供两种硬编码矩阵:
IUB。这是BESTFIT用于比较核酸序列的默认评分矩阵。 X和N被视为与任何IUB模糊度符号的匹配。所有比赛得分1.0; IUB符号的所有不匹配得分均为0.9。
CLUSTALW(1.6)。 ClustalW使用的先前系统,其中匹配项得分为1.0,不匹配项得分为0。IUB符号的所有匹配项也得分为0。
如果文件名仅包含小写字符,则可以从磁盘上的文件读取新矩阵。新的权重矩阵中的值应该相似并且对于不频繁的替换应该为负。
输入格式。用于新矩阵的格式与BLAST程序相同。任何以“#”字符开头的行均被视为注释。第一个非注释行应包含一个任意顺序的氨基酸列表,使用1个字母代码,后跟一个“ *”字符。然后是分数的方阵,每个氨基酸一行一行,一列。矩阵的最后一行和最后一列(对应于“ *”字符)包含整个矩阵的最小分数。
质量得分参数
您可以使用以下选项来自定义在对齐显示下方绘制的“质量得分”列。
SCORE PLOT SCALE:这是一个从1到10的标量值,可用于更改质量得分图的比例。
残渣排除:这是一个标量值,范围是1到10,可用于更改在比对显示中突出显示的残渣例外的数量。 (有关此临界值的说明,请参见下面的“残留物的计算”部分。)
蛋白质重量矩阵:评分表,描述每种氨基酸之间的相似性。
DNA权重矩阵:提供两种硬编码矩阵:IUB和CLUSTALW(1.6)。
显示多余的残留物
此选项突出显示在比对质量计算中得分很差的单个残基。得分极低的残基通过在灰色背景上使用白色字符突出显示。
保存质量分数到文件
对齐显示下方绘制的质量得分也可以保存在文本文件中。路线中的每一列都写在输出文件中的一行上,质量得分的值在该行的末尾。仅将当前在显示屏中选择的序列写入文件。质量得分的一种用途是通过序列保守来着色蛋白质结构中的残基。这样,可以突出保守的表面残基以定位功能区,例如配体结合位点。
质量得分的计算
假设我们有m个长度为n的序列的比对。然后,对齐方式可以写为:
A11 A12 A13 ..... A1n
A21 A22 A23 ..... A2n
。
。
Am1 Am2 Am3 ..... Amn
我们还有一个大小为R的残基比较矩阵,其中C(i,j)是将残基i与残基j对齐的分数。
我们要计算比对中第j个位置的保守性得分。
为此,我们定义了R维序列空间。对于比对中的第j个位置,每个序列都由一个残基组成,该残基在空间中分配了一个点S。 S具有R维,对于序列i,第r维定义为:
Sr = C(r,Aij)
然后,我们为比对中的第j个位置计算一个共识值。该值X也具有R维,第r维定义为:
Xr =(SUM(Fij * C(i,r)))/ m
1 <= i <= R
其中Fij是比对中第j位的残基i的计数。
现在我们可以计算每个序列i与R维空间中的共识位置X之间的距离Di。
Di = SQRT(SUM(Xr-Sr)(Xr-Sr))
1 <= i <= R
将比对中第j个位置的质量得分定义为序列距离Di的平均值。
通过乘以在该位置具有残基(而不是缺口)的序列的百分比来对分数进行归一化。
残留物的计算
如果序列与共识值P的距离Di大于(四分位数上限+四分位数间距*截止值),则第i个序列的第j个残基被视为例外。可以从SCORE PARAMETERS菜单中设置用作显示异常的临界值。较高的截止值将仅显示非常重要的异常。较低的值将突出显示更多,不太重要的异常。
(注意:在此位置包含缺口的序列不包括在异常计算中。)
低分段的计算
假设我们有m个长度为n的序列的比对。然后,对齐方式可以写为:
A11 A12 A13 ..... A1n
A21 A22 A23 ..... A2n
。
。
Am1 Am2 Am3 ..... Amn
我们还有一个大小为R的残基比较矩阵,其中C(i,j)是将残基i与残基j对齐的分数。
我们通过建立一个邻接树来计算序列权重,其中分支长度与散度成正比。通过分支所有权对分支求和可得出权重。参见(Thompson等,CABIOS,10,19(1994)和Henikoff等,JMB,243,574,1994)。
为了找到序列Si中的低得分片段,我们建立了比对中其余序列的加权图。假设我们在序列的第j位找到了残基r;然后将序列中第j个位置的分数定义为
分数(Si,j)=轮廓(j,r),其中轮廓(j,r)是轮廓分数
对于在位置j处的残基r
对准。
这些残差得分沿着序列在向前和向后两个方向上求和。如果分数总和为正,则将其重置为零。在两个方向上均得分为负的线段被视为“低得分”,并将在对齐显示中突出显示。
人气软件
-

endnote x9.1中文版下载 107.0 MB
/简体中文 -

Canon IJ Scan Utility(多功能扫描仪管理工具) 61.55 MB
/英文 -

A+客户端(房源管理系统) 49.6 MB
/简体中文 -

第二代居民身份证读卡软件 4.25 MB
/简体中文 -

船讯网船舶动态查询系统 0 MB
/简体中文 -

ZennoPoster(自动化脚本采集/注册/发布工具) 596.65 MB
/英文 -

中兴zte td lte 18.9 MB
/简体中文 -

originpro 2021 527 MB
/英文 -

个人信息管理软件(AllMyNotes Organizer) 5.23 MB
/简体中文 -

ZKTeco居民身份证阅读软件 76.2 MB
/简体中文
 有道云笔记 8.0.70
有道云笔记 8.0.70  Efficient Efficcess Pro(个人信息管理软件) v5.60.555 免费版
Efficient Efficcess Pro(个人信息管理软件) v5.60.555 免费版  originpro8中文 附安装教程
originpro8中文 附安装教程  鸿飞日记本 2009
鸿飞日记本 2009  竞价批量查排名 v2020.7.15 官方版
竞价批量查排名 v2020.7.15 官方版  Scratchboard(信息组织管理软件) v30.0
Scratchboard(信息组织管理软件) v30.0  Fitness Manager(俱乐部管理软件) v9.9.9.0
Fitness Manager(俱乐部管理软件) v9.9.9.0 









