

MEGA(分子进化遗传分析软件)
v7.0.26 官方版- 软件大小:30.8 MB
- 更新日期:2020-08-11 14:55
- 软件语言:英文
- 软件类别:理科工具
- 软件授权:免费版
- 软件官网:待审核
- 适用平台:WinXP, Win7, Win8, Win10, WinAll
- 软件厂商:

软件介绍 人气软件 下载地址
MEGA提供遗传分析功能,软件提供丰富的分析方案,支持比对序列、估计进化距离、从序列数据构建树、测试树的可靠性、处理基因和域、选择测试、与组一起管理分类单元、计算序列统计,软件界面显示功能菜单,点击对应的菜单就可以弹出分析界面,将您的数据加载到软件就可以执行分析,支持模式异质性的差异指数检验,支持估算过渡/过渡偏差,也可以选择计算MCL转换/转换偏差、计算模式差异指数、计算构图距离,大部分功能都提供详细的帮助操作,用户可以在帮助界面查看官方提供的教程文件,从而了解这款软件如何分析实验数据!
新版功能
版本7的新功能
版本7对MEGA6进行了许多增强。它们包括
针对64位处理器体系结构的优化源代码。 MEGA现在可以使用许多GB的内存(取决于可用的硬件资源和操作系统),而MEGA以前只能使用<4GB的RAM。 (注意*** MEGA7和将来的版本将不会为32位体系结构分发)。
添加了用于识别树中基因复制事件的向导式系统。另外,如果提供了物种树,将使用Zmasek和Eddy(2001)的算法识别物种形成事件。
重构了“树资源管理器”,以便可以显示具有多达10万个分类单元的树。这取决于可用的内存和图形处理资源。
重构时间树系统,以估计系统发育中所有分支点的相对和绝对发散时间(Tamura等,2012),以使用更直观的向导式界面。
更新了树浏览器,以显示推断树中所有内部节点的数据覆盖率。数据覆盖率值可用于识别树中有问题的节点,由于数据覆盖率不足,其分支长度估计可能不可靠。
现在,“时间树”系统要求所有时间树都使用输出组生根。用户界面已更新了两种用于指定外组的方法(通过“树资源管理器”或通过“分类/组”对话框)。
添加了用于导出树图像的其他图像格式。
对ImageMagick的依赖性已被删除。
Caption Expert系统已更新,因此字幕可以停靠在Tree Explorer窗口中
软件特色
文本文件编辑器和格式转换器
MEGA包含一个文本文件编辑器,可用于创建和编辑ASCII文本文件。如果输入数据文件处理模块检测到数据文件格式的错误,则由MEGA自动调用。在这种情况下,您应进行适当的更改并保存数据文件。
Alignment Explorer
Alignment Explorer提供以下选项:(1)查看和手动编辑对齐;(2)使用内置CLUSTALW实现和MUSCLE程序生成对齐(对于任何矩形区域中的完整序列或数据)。 Alignment Explorer还提供了用于探索基于Web的数据库(例如,NCBI查询和BLAST搜索)并将所需序列数据直接检索到当前对齐的工具。
关于MUSCLE
MUSCLE是用于产生氨基酸和核苷酸序列的多重比对的程序。 将MUSCLE的速度和准确性与T-Coffee,MAFFT和CLUSTALW进行比较,并在所有测试中达到最高或联合最高等级。 如果在不进行细化的情况下使用,其精度与T-Coffee或MAFFT相同,并且在调整三者中的大序列时最快。
关于CLUSTALW
ClustalW是广泛使用的用于比对任何数量的同源核苷酸或蛋白质序列的系统。对于多序列比对,ClustalW使用渐进比对方法。在这些中,最相似的序列,即具有最佳比对分数的序列首先对齐。然后逐渐更远的序列组对齐,直到获得全局对齐。这种启发式方法是必要的,因为找到全局最优解决方案在内存和时间要求方面都是过高的。
安装方法
1、打开MEGA7.0.26_win64_setup.exe软件直接安装,点击下一步
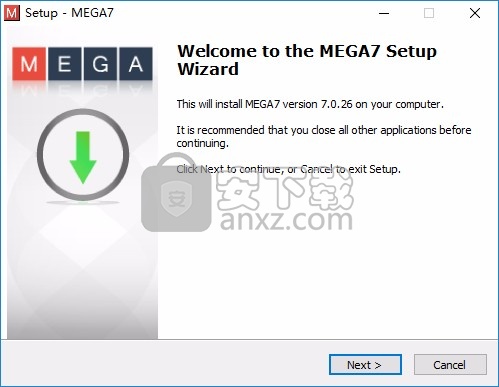
2、设置软件的安装地址C:\Program Files\MEGA7
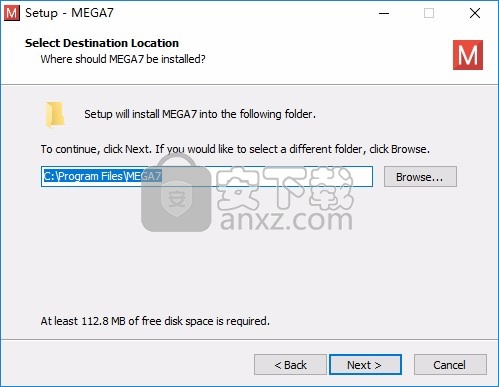
3、提示软件的安装快捷方式名字
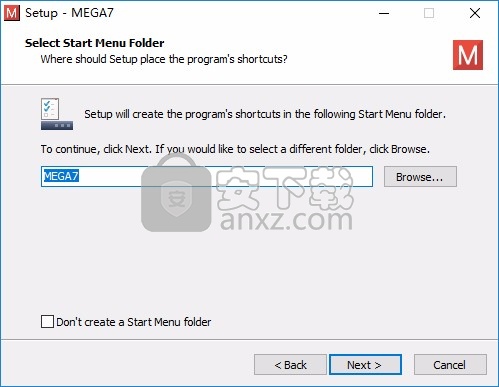
4、提示软件的启动图标设置界面,勾选界面的选项
5、提示关联的格式,默认第一个
6、提示安装信息查看,你设置的内容都在这里显示
7、提示安装进度界面,等待软件安装完毕吧
8、MEGA7成功安装到电脑,点击完成
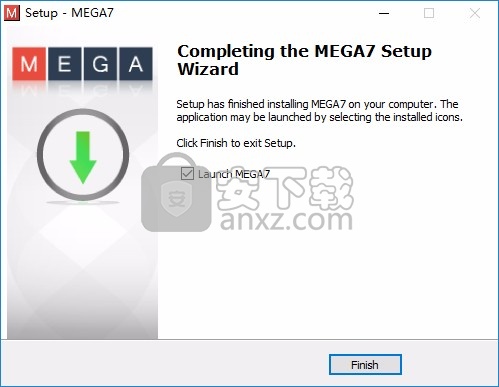
9、打开主程序就可以正常使用,可以的顶部查看分析菜单功能
官方教程
演练简介
本演练提供了一些简短的教程,解释了如何在MEGA中执行常见任务。每个教程都需要使用示例数据文件,该文件可以在/ MEGA / Examples文件夹中找到(Windows用户的默认位置为C:\ Users \ UserName \ Documents \ MEGA7 \ Examples \。Mac用户的位置为$ HOME / MEGA / Examples,其中$ HOME是用户的主目录)。建议您按照给出的顺序按照给定教程的示例进行操作,因为初始示例中说明的技术将在后续示例中再次使用。
在教程中,使用以下约定:
按键由粗体字母表示(例如F4)。
如果必须同时按下两个键,则它们之间会显示一个+号(例如Alt + F3表示应同时按下Alt和F3键)。
斜体字表示菜单或窗口的名称。
斜体粗体字表示在菜单,子菜单和工具栏中找到的单个命令。
“主菜单”指的是当前活动窗口(文件,分析,帮助等)顶部的菜单栏。
“ MEGA主菜单”是指您在MEGA主窗口中启动所有分析的菜单。
“启动栏”是指直接位于当前活动窗口的主菜单(“对齐”,“数据”,“模型”,“距离”等)下方的工具栏

为简便起见,菜单/按钮的单击顺序由一系列用管道分隔的命令指示(例如,“文件|打开”表示您应单击“文件”主菜单项,然后单击“打开”子菜单) 显示的菜单项)。
比对序列
在本教程中,我们将展示如何从蛋白质序列数据创建多序列比对,该数据将使用不同的方法导入比对编辑器。可以在MEGA \ Examples \文件夹中找到本教程中使用的所有数据文件(Windows用户的默认位置为C:\ Users \ UserName \ Documents \ MEGA7 \ Examples \\。Mac用户的位置为$ HOME。 / MEGA / Examples,其中$ HOME是用户的主目录)。
打开路线
Alignment Explorer是用于在MEGA中构建和编辑多个序列比对的工具。
范例2.1:
通过选择Align |启动Alignment Explorer。在MEGA主窗口的启动栏上编辑/生成路线。
选择“创建新路线”,然后单击“确定”。将出现一个对话框,询问“您是否正在建立DNA或蛋白质序列比对?”点击标有“ DNA”的按钮。
在Alignment Explorer主菜单中,选择Data |开启|从文件中检索序列。从MEG / Examples目录中选择“ hsp20.fas”文件。
通过ClustalW进行序列比对
您可以使用ClustalW或Muscle算法在MEGA中创建多序列比对。在这里,我们使用ClustalW选项比对一组序列。
范例2.2:
打开对齐文件(使用上面的说明)hsp20.fas。
选择编辑| Select All菜单命令选择数据集中每个序列的所有位点。
选择对齐|在主菜单中按ClustalW进行比对,以使用ClustalW算法比对选定的序列数据。单击“确定”按钮接受ClustalW的默认设置。
对齐完成后,通过选择数据|保存当前对齐会话。从主菜单中保存会话。为文件指定一个适当的名称,例如“ hsp20_Test.mas”。这将允许还原当前的对齐会话以供将来编辑。
通过选择数据|退出对齐资源管理器。从主菜单退出Aln Explorer。
使用肌肉比对序列
在这里,我们描述了如何使用Muscle选项创建多序列比对。
范例2.3:
从MEGA主窗口开始,选择Align |。从启动栏中编辑/构建路线。选择创建新的比对,然后选择DNA。
在Alignment Explorer窗口中,选择Data |开启|从文件中检索序列,然后从MEGA / Examples目录中选择“ Chloroplast_Martin.meg”文件。
在Alignment Explorer主菜单上,选择Edit |全选。
在Alignment Explorer启动栏上,您将找到一个看起来像屈臂的图标。单击它,然后选择“比对DNA”。
在MUSCLE-AppLink窗口的底部附近,您将看到名为Alignment Info的行。您可以阅读有关Muscle程序的信息。
单击计算按钮(接受默认设置)。 “进度”窗口将使您了解肌肉对齐状态。在此窗口中,您可以单击“命令行输出”选项卡以查看传递给Muscle程序的命令行参数。注意:分析可能会很快完成,您将无法单击该标签或阅读该标签。此标签中的信息不是必不可少的,只是很有趣。
当Muscle程序完成时,对齐的序列将被传递回MEGA,并显示在Alignment Explorer窗口中。
通过选择数据|关闭对齐资源管理器。退出Aln Explorer。当询问您是否要将当前对齐会话保存到文件时,选择否。
从Internet(GenBank)获取序列数据
如果您的互联网连接有效,则可以使用MEGA的集成网络浏览器从NCBI网站获取GenBank序列数据。
范例2.4:
在MEGA主窗口中,选择Align |。从主菜单中编辑/构建路线。
出现提示时,选择“创建新路线”,然后单击“确定”。选择DNA
通过选择网络|激活MEGA的集成浏览器。从主菜单中查询Genbank。
加载NCBI:核苷酸网站后,将CFS作为搜索词输入到屏幕顶部的搜索框中。按搜索按钮。
显示搜索结果时,选中要导入到MEGA中的任何项目旁边的框。
如果您选中了多个框,则:找到“显示设置”下拉菜单(位于页面左上角的选项卡标题正下方)。将值更改为FASTA(文本),然后单击“应用”按钮。这将以FASTA格式输出您选择的所有序列作为文本。
按下网址栏上方的添加到对齐按钮(带有红色+号)。这会将序列导入比对资源管理器。
现在,数据显示在Alignment Explorer中,您可以关闭Web Browser窗口。
使用前面的示例中详细介绍的步骤对齐新数据。
单击“数据” |“关闭”,然后单击“路线”。退出Aln Explorer。当询问您是否要将当前对齐会话保存到文件时,请选择“否”。
注意:我们已经比对了一些序列,现在可以对其进行分析了。每当需要编辑/更改序列数据时,都需要在“比对编辑器”中将其打开,然后在此处进行编辑或比对。然后将其导出为MEGA格式并打开生成的文件。
估计进化距离
在本教程中,我们将使用各种模型来估计11种果蝇序列的进化距离。可以在MEGA / Examples文件夹中找到本教程中使用的数据文件(Windows用户的默认位置为C:\ Users \ UserName \ Documents \ MEGA7 \ Examples \。Mac用户的默认位置为$ HOME / MEGA /示例,其中$ HOME是用户的主目录)。
使用成对距离估计进化距离
在MEGA中,您可以通过计算每对序列之间核苷酸差异的比例来估计序列之间的进化距离。
范例3.1:
打开“ Drosophila_Adh.meg”数据文件。如果需要,请参阅“ MEGA基础”教程。
在主要的MEGA启动栏中,选择Distance |计算成对距离。
在“分析首选项”窗口中,单击“取代类型”下拉列表,然后选择“核苷酸”选项。
单击“模型/方法”下拉菜单,然后选择p距离模型。在此示例中,我们将使用默认值作为其余选项。单击计算开始计算。
进度指示器将短暂出现,然后距离计算结果将以网格形式显示在新窗口中。将此窗口保持打开状态,以便我们可以比较后续步骤的结果。
使用其他模型/方法计算和比较距离
MEGA支持用于估计进化距离的大量模型。在这里,我们比较使用不同模型计算出的进化距离。
范例3.2:
重复上面的示例3.1,但在“模型/方法”下拉列表中选择“ Jukes /康托尔”模型,而不是p距离模型,其他所有选项均保持不变。同样,将结果窗口保持打开状态以进行比较。
重复分析,这次在“模型/方法”下拉菜单中选择“田村-Nei”模型,所有其他选项保持不变。同样,将结果窗口保持打开状态以进行比较。
现在,您可以比较三个打开的结果窗口,其中包含通过不同方法估算的距离。
比较结果后,选择文件|退出每个结果窗口的查看器选项。不要关闭“ Drosophila_Adh.meg”数据文件。
计算氨基酸差异的比例
您还可以根据氨基酸差异的比例计算进化距离。
注意:MEGA将使用选定的遗传密码表将核苷酸序列自动翻译为氨基酸序列。遗传密码表可以通过Data |从主MEGA启动栏中选择“遗传密码表”。
范例3.3:
在MEGA主窗口中,选择Distance |从主菜单计算成对距离。这将显示“分析首选项”窗口。
单击“替代类型”下拉列表,选择“氨基酸”,然后在“模型/方法”下选择“ p-距离”。
单击“计算”按钮以接受其余选项的默认值,然后开始计算。进度对话框将短暂出现。与核苷酸估计一样,将显示结果查看器窗口,以网格格式显示距离。
检查结果后,使用“文件” |“文件”。退出查看器命令以关闭结果查看器。
通过选择主MEGA任务栏上的“关闭数据”按钮来关闭数据。
从序列数据构建树
在本教程中,我们将使用“数据”和“系统发育”菜单中的可用命令来说明构建树和内存中序列数据的过程。我们将使用MEGA / Examples目录中的“ Crab_rRNA.meg”文件。该文件包含来自不同蟹类的大亚基线粒体rRNA基因的核苷酸序列(Cunningham等,1992)。由于rRNA基因被转录但未被翻译,因此属于非编码基因。
可在MEGA / Examples文件夹中找到本教程中使用的“ Crab_rRNA.meg”文件(Windows用户的默认位置为C:\ Users \ UserName \ Documents \ MEGA7 \ Examples \。Mac用户的默认位置为$ HOME / MEGA / Examples,其中$ HOME是用户的主目录)。
建立邻居加入(NJ)树
在此示例中,我们将说明使用MEGA重建系统树的基础知识,并熟悉“树资源管理器”窗口。
范例4.1:
激活“ Crab_rRNA.meg”数据文件。如有必要,请参阅“ MEGA基础”教程的示例1.2。
在主要的MEGA启动栏中,选择系统发生| “构造/测试邻居加入树”菜单选项。
在“分析首选项”窗口中,从“模型/方法”下拉列表中选择p距离选项。
单击“计算”以接受其余选项的默认值,然后开始计算。在树显示在“树资源管理器”窗口中之前,进度指示器将短暂出现。
要选择一个分支,请用鼠标左键单击它。如果使用鼠标右键单击分支,则会出现一个小的选项菜单,您可以通过该菜单翻转分支并对其执行各种其他操作。
选择一个分支,然后按向上,向下,向左和向右箭头键查看光标如何在树中移动。
通过选择视图|更改分支样式。 “树资源管理器”主菜单中的“树/分支样式”命令。
选择视图|从“树资源管理器”主菜单中选择“仅拓扑”命令,以在屏幕上显示分支模式。
您可以通过选择“视图” |“仅拓扑”选项来显示数字分支长度。单击选项,然后单击分支选项卡。选中标有“显示分支长度”的框,然后单击“确定”。
打印NJ树(适用于Windows用户)
Windows用户可以直接从Tree Explorer中打印。
范例4.2a:
选择文件|从“树资源管理器”主菜单中选择“打印”选项,以显示一个标准的“打印”窗口。这将以全尺寸打印树,并可能需要多张纸。按取消。
要将打印树的大小限制为单张纸,请选择“文件” |“文件”。从“树资源管理器”主菜单中的“图纸”命令中打印。按确定。
选择文件|退出树浏览器命令以退出树浏览器。单击确定按钮关闭树浏览器而不保存树会话。
打印NJ树(对于Mac用户)
在Mac系统上运行时,MEGA不支持直接从Tree Explorer进行打印。要在Mac上打印树,用户可以将树图像保存为PDF文件,然后以常规方式打印。
范例4.2b:
选择图像|从“树资源管理器”主菜单中的“另存为PDF文件”选项,以调出标准的“保存”窗口。将图像保存到所需位置。
保存文档后,您可以使用PDF阅读器打开它,并以与其他任何PDF文档相同的方式打印文档。
选择文件|退出树浏览器命令以退出树浏览器。单击确定按钮关闭树浏览器而不保存树会话。
使用分支界限搜索选项构造最大简约(MP)树
使用MEGA,除了邻居加入之外,您还可以使用最大似然,最小进化,UPGMA和最大简约方法来重建系统发育。在这里,我们使用最大简约(MP)方法重建“ Crab_rRNA.meg”数据的系统发育。
范例4.3
选择系统发育|从主MEGA启动栏构建/测试最大简约树(S)菜单选项。在“分析首选项”窗口中,为“ MP搜索方法”选项选择“最大-最小分支限界”。
单击“计算”按钮接受其他选项的默认值,然后开始计算。进度窗口将短暂出现,并且该树将显示在“树资源管理器”中。
(Windows用户)现在通过从“树资源管理器”的“文件”菜单中选择“打印”选项来打印该树。
(Mac用户)按照上面的示例4.2b中所述将树保存到PDF文件。
比较NJ和MP树。对于此数据集,这两个树的分支模式是相同的。
选择文件|退出树浏览器命令以退出树浏览器。单击确定关闭树浏览器,而不保存树会话。
使用启发式搜索构造MP树
对于每种系统发育推断方法,MEGA提供了许多选择。在此示例中,我们使用Min-Mini Heuristic搜索进行MP分析。
范例4.4:
请按照示例4.3中的步骤操作,而不是选择“最大-最小”分支约束,而是选择“最小-最小启发式”作为MP搜索方法。将MP搜索级别更改为2,然后单击计算。
注意:在此示例中,只要MP Search Level设置为2,就可以通过Max-mini Branch-&-bound选项获得与Min-Mini Heuristic选项相同的树。但是,计算时间要短得多启发式方法。
检查数据编辑功能
对于非编码序列数据,可以选择OTU(操作分类单位)以及位点进行分析。
范例4.5:
在MEGA主窗口中,选择Data |从启动栏中选择“分类单元和组”选项。显示对话框。
在左侧面板中检查了所有OTU标签。这表明所有OTU都包含在当前活动数据子集中。要从数据中删除第一个OTU,请取消选中左侧面板中第一个OTU名称旁边的复选框。单击关闭按钮。
现在,当您从此数据集构造邻居合并的树时,它将包含12个OTU而不是13个OTU。退出树浏览器,不保存。通过从MEGA主窗口中选择“关闭数据”图标来停用操作数据集。
人气软件
-

MathType(数学公式编辑器) 9.9 MB
/简体中文 -

MathType 7.1注册工具 38.4 MB
/简体中文 -

经纬度距离角度计算器 12.7 MB
/简体中文 -

Mathtype 金山公式编辑器 41.00 MB
/简体中文 -

小学数学出题软件 0.02 MB
/简体中文 -

动态几何软件 GeoGebra 6.0.472 中文版 48.00 MB
/简体中文 -
ZX数学函数作图器 1.16 MB
/简体中文 -
MEGA(分子进化遗传分析软件) 30.8 MB
/英文 -

小学数学试卷生成器 17.6 MB
/简体中文 -
分解质因数 2.04 注册版 2.00 MB
/简体中文
 geogebra(几何画板) v6.0.794.0
geogebra(几何画板) v6.0.794.0  GeoGebra(动态数学软件) 6.0.790.0
GeoGebra(动态数学软件) 6.0.790.0  maplesoft maple 2020中文(可视化分析与探索) 附安装教程
maplesoft maple 2020中文(可视化分析与探索) 附安装教程  HyperChem补丁 附带安装教程及序列号
HyperChem补丁 附带安装教程及序列号 














